Transcriptome-wide Screening in Cell Culture with SLALOM and lentiCRISPRv2
Published on: September 14, 2022
Updated on: September 14, 2022
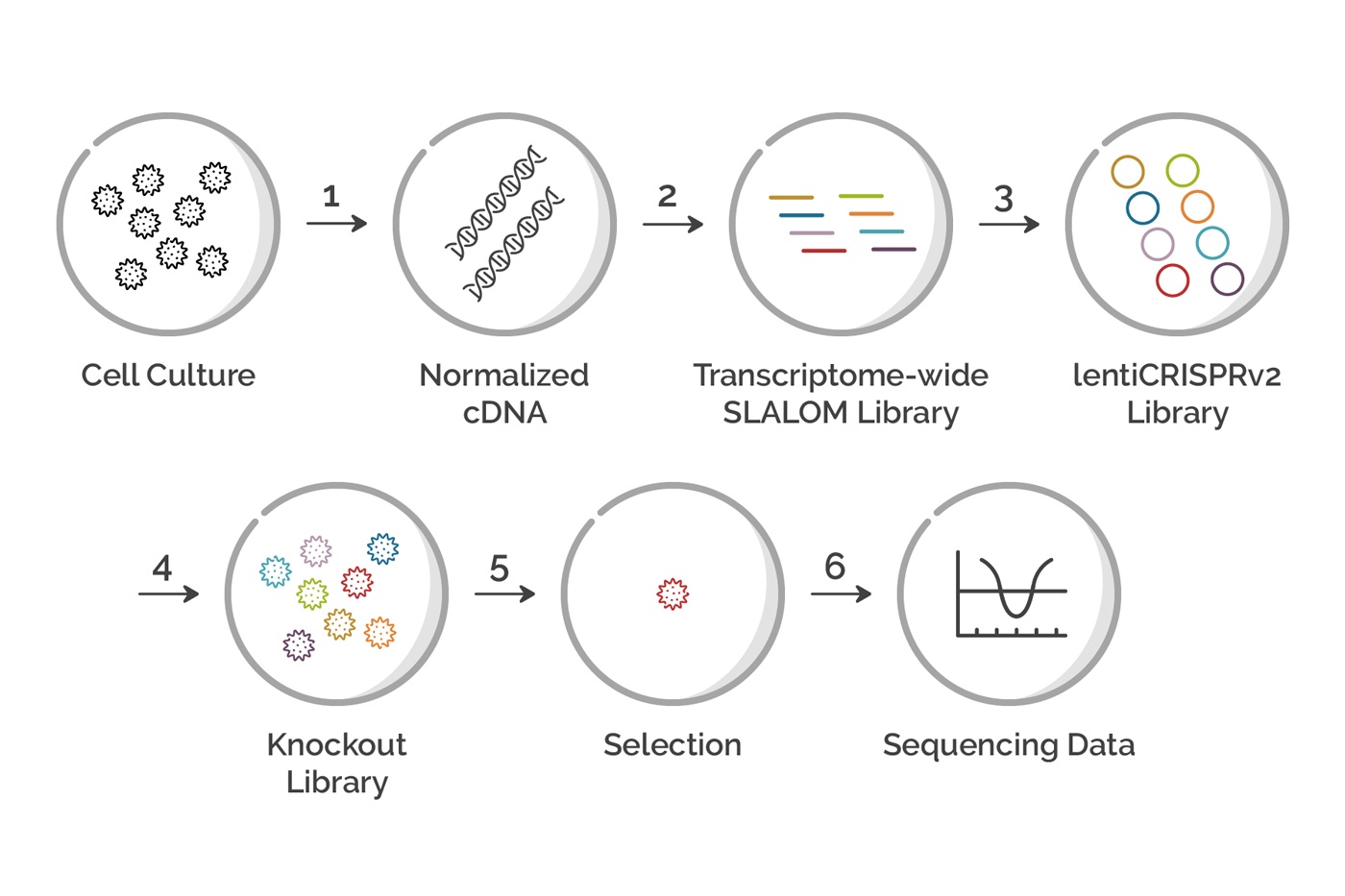
Figure 1: Flowchart showing each step in a Genome-wide CRIPSR Screen. Each step in the process is described in detail in the article below.
Conducting a forward-genetic screen can seem like a daunting task, but it doesn’t have to be. Recent developments in CRISPR technology have made it easier than ever to introduce mutations into any cell line. With the SLALOM 1.0 ™ sgRNA library generation kit, it is now possible to make sure the libraries are variant-matched and customized to your specific application. Together, these technologies will allow novel genes and pathways to be identified faster than ever before.
CRISPR Screening in Cell Culture
The CRISPR/Cas9 system is a two-component system consisting of a DNA nuclease and a single-guide RNA (sgRNA). By simply modifying the nucleotide sequence of the sgRNA, it can be programmed to cleave at different sites in the genome, creating insertions or deletions that disrupt the coding sequence of a protein. The phenotype caused by the mutation can then be used to study the function of the gene.
The CRISPR/Cas9 system makes it simple to knockout individual genes, but it can also be used at the genome scale to identify relevant genes and pathways. The first step in a genome-wide CRISPR screen is the synthesis of a pooled sgRNA library targeting all genes in a genome to create a population of cells where each of the cells has a different gene knocked out. A selective pressure is then applied to the population and sgRNAs corresponding to the phenotype of interest are subsequently sequenced to identify a comprehensive set of the genes involved in the process being studied. Thus a high quality sgRNA library targeting the genes is essential for getting good results in a screen.
A few genome-wide sgRNA libraries have been designed and chemically synthesized that target the coding regions for a few annotated genomes. However, there are many situations where a custom library is needed or can provide better results. The synthesis of a custom sgRNA library using traditional microarray technology is expensive and often requires over a month to design and have synthesized. In addition, these libraries aren’t completely accurate because their sgRNAs are still designed using a reference genome.
SLALOM 1.0 ™ kits solve these problems by enzymatically capturing many possible sgRNAs directly from the protein-coding sequences of your specific cell line, producing a truly variant-matched library. And, it will be ready to use in less than one day for a fraction of the cost. Using SLALOM 1.0 ™ kits to create libraries targeting specific subsets of genes by using mRNA (all expressed genes), ChIP pulldowns (genes with particular histone marks), or PCR products (specific regions of the genome) will help researchers more efficiently identify candidate genes, as focuses on genetic loci that are more likely to be of interest and increases coverage of those genes.
In this article we walk through each step of a transcriptome-wide CRISPR screen using SLALOM 1.0 ™.
Steps in a Genome-wide CRIPSR Screen
While each CRISPR screen is unique, they all share a common set of steps. Here, we will walk you through the steps for a design using a SLALOM 1.0 ™ gRNA library made from a total mRNA prep. The process outlined here can be easily applied to other similar designs with minor changes.
Step 1 - Generate Normalized cDNA
In this example, we are making a library targeting the coding regions of genes expressed in the cell line we are studying. To prepare the samples for sgRNA library creation, we must first extract the RNA, convert it to a double-stranded cDNA library, and normalize the DNA. RNA can be extracted from cells using a wide range of available kits and follows the same procedure you would use for qPCR, RNA-seq, or any other downstream application.
Following RNA extraction, the next step is to convert it to cDNA. While most kits only make single-stranded cDNA, we need to make double-stranded cDNA to allow for library normalization. We recommend using the Evrogen Mint-2 cDNA synthesis kit. This kit will use a polyA tail specific primer. Thus, it will automatically remove ribosomal RNAs and other non-polyadenylated species.
The amount of each gene in the cDNA library is proportional to the expression level of the gene in the cells. As some genes are highly expressed, our sgRNA library would be dominated by guides targeting a handful of genes. To prevent this, we must normalize the cDNA to even out the representation of the genes. cDNA normalization is a standard process used in high-throughput sequencing, so several kits exist. One such kit is the Evrogen Trimmer-2 cDNA normalization kit. In this kit, the cDNA is heated to separate the forward and reverse strands. It is then quickly cooled and digested with a double-stranded DNA nuclease. The probability of a strand finding its partner is proportional to its concentration in the mix. Thus, highly expressed genes are more likely to hybridize to form double stranded DNA, which is then digested. In one of those beautiful mathematical symmetries, the result is an even representation of all genes in the resulting pool. The DNA is now ready to use to make an sgRNA library.
Step 2 - Make the sgRNA library
The normalized cDNA can be enzymatically converted into a Genome-wide sgRNA library using the SLALOM 1.0 ™ sgRNA Library Synthesis Kit (Pioneer Biolabs). The entire procedure takes less than a day and can be performed on the bench using basic lab equipment. A complete protocol can be found here [link], and kits can be found here [link]. The kit produces high-quality, variant-matched sgRNA libraries targeting, in our case, the entire expressed transcriptome of the cells at a density of about 2 sgRNAs (both forward and reverse strands) every 200 bp of the genome.
Step 3 - Clone into the LentiCRISPRv2 plasmid
The SLALOM 1.0 ™ kit comes in several variants. The LentiCRISPRv2 option contains the restriction sites necessary to clone the sgRNAs into the plasmid. Thus, cloning is as simple as a digest with Esp3I, ligation, and transformation in E. Coli. As transforming a complete library would create a lot of colonies to pick, we have found it is better to use a pooled approach where the transformed bacteria are directly grown in liquid culture. In this method, bacteria are mixed with the plasmid and electroporated to induce plasmid uptake. The cells are then grown in a rich medium for 1 hour. Following the initial recovery incubation, 90% of the culture is added to a 5 ml culture overnight. The remaining 10% is plated to estimate the total number of colonies. The following day, the culture can be miniprepped to create a pooled mixture of plasmids representing the library. It is important to ensure that you are cloning enough sgRNAs to represent the library. We recommend at least 10x the estimated size of your library, which may take multiple electroporations to accomplish.
Step 4 - Lentiviral Packaging and Transduction of Cells
The LentiCRISPRv2 plasmid is designed to express both Cas9 and sgRNAs from a standard 3rd generation lentivirus backbone. Thus, lentivirus can be generated at this point using common protocols. Briefly, LentiCRISPRv2 plasmid library is co-transfected into Hek293 cells with two helper plasmids: pMD2.G and psPAX. The cells are then grown for 2 days, and then the virus is harvested and filtered from the media. After measuring the viral titer of the media, the cells to be screened are transduced with the lentivirus at an MOI < 0.3. This low multiplicity of infection ensures that the cells do not take up more than one sgRNA, as this can confound results.
Step 5 - Screening and Analysis
There are many ways to isolate cells with the phenotype of interest and to identify the genes associated with the phenotype. Here, we will cover the two most commonly used methods in cell cultures.
The Enrichment Method
The enrichment method can be used any time you are looking for mutations that affect cell viability or division. Such phenotypes include resistance/susceptibility to drugs or chemicals, cell division signaling pathways, the ability to metabolize certain chemicals, etc. In this method, the library is introduced to the cells and expression of the sgRNA and Cas9 are induced. If you are interested in viability genes, then an uninduced culture is kept as a control. In most other cases, you will induce in the presence and absence of the condition you are testing to create a standard case-control comparison. Both cells are cultured for several generations and then collected. The sequences of the gRNAs are then PCR amplified from the genome and analyzed using high-throughput sequencing. If a gene targeted by a particular gRNA makes the cells more susceptible to the treatment, then that gRNA will be underrepresented in the treated cells compared to control. If it makes the cells more resistant, then it will be overrepresented in the treatment group compared to control. Of note, comparison between the case and control is essential, as you cannot assume the gRNAs were evenly represented in the original population. We also recommend running several replicates in parallel, as this will help differentiate between enrichment/depletion and random genetic drift in the population.
The Sorting Method
After sequencing some of the colonies on the large plates, it becomes apparent that there are actually multiple enzymes involved in the metabolism of substrate S. If this is the case, picking and sequencing individual colonies could quickly become unwieldy. Fortunately, the color difference between the cells that metabolize substrate S and those that do not can be used to sort the cells into two subpopulations using Fluorescence-activated Cell Sorting (FACS). The bacterial repression library is grown in liquid broth containing IPTG and the cells are sorted based on color. Genomic DNA from the white cells is then purified and the sgRNA sequences are PCR amplified and sent off for high through-put sequencing. The genes are then aligned back to the genome to determine candidate genes involved in the metabolism of substrate S.
The Enrichment Method
If no lethal phenotype can be identified, then the best method is to create a system where the cells can be separated into two populations based on a visual marker. For example, fluorescent proteins or FRET probes can be used to measure the level of activity or changing conditions in a particular cell. These cells can then be transfected, incubated for a certain amount of time, and then sorted based on fluorescence into phenotypic and non-phenotypic populations. The sequences of the gRNAs are then PCR amplified from the genome and analyzed using high-throughput sequencing. If a gene targeted by a particular gRNA results in a phenotype, then that gRNA will be overrepresented in the phenotypic cells compared to the non-phenotypic cells. Of note, comparison between the two populations or to a control is essential, as you cannot assume the gRNAs were evenly represented in the original population. We also recommend running several replicates in parallel, as this will help differentiate between enrichment/depletion and random genetic drift in the population.
Summary
Thanks to CRISPR and SLALOM 1.0 ™ technology, forward-genetic screens are more accessible than ever before. Using these techniques, a screen can be completed in a matter of weeks for a fraction of the cost. There has never been a better time to discover the genes driving your process of interest. As always, if you need help setting up your experiment, we are standing by to consult and help you see how CRISPR screening can fit in your gene discovery workflow.
Have additional questions or want to talk about designing a genome-wide CRISPRi screen? Please feel free to reach out to us to schedule a free consulting appointment at: support@pioneerbiolabs.com
References
1. Yates JD, Russell RC, Barton NJ, Yost HJ, Hill JT. A simple and rapid method for enzymatic synthesis of CRISPR-Cas9 sgRNA libraries. Nucleic Acids Res. 2021;49(22):e131.
2. Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11(8):783-784.